Cracking the code of Huntington's disease
/iStock photos (human sketch)")
The structure of huntingtin (EBI)/iStock photos (human sketch)
Huntington’s disease is caused by a gene mutation that causes a protein to build up in the brain. In a world first, EPFL scientists have synthesized and studied modified forms of a mutant part of the protein, deepening our understanding of how it contributes to the disease, and pointing to new therapeutic strategies for treating it.
Huntington’s disease is a neurodegenerative disorder that causes patients to lose their ability to move, speak, and even think. It is caused by a gene mutation that produces an abnormal form of the protein huntingtin, which aggregates and builds up inside neurons of the cortex and striatum. Small chemical modifications on different parts of huntingtin could reduce its toxicity and aggregation, but as many enzymes already chemically modify the protein in the cell, it has been difficult to determine what chemical modifications could serve as future therapies. EPFL scientists have now developed synthetic methods that allow site-specific chemical modifications on huntingtin while bypassing the need to identify the enzymes behind them. The study is published in Angewandte Chemie.
After being produced by its gene, huntingtin is subjected to numerous chemical changes, during which enzymes in the cell attach different chemical groups to it, such as phosphate (phosphorylation) or acetylene (acetylation). These changes are called “post-translational modifications” but the key molecular players that regulate them remain unknown.
To address this knowledge gap and explore the therapeutic potential of post-translational modifications, Hilal Lashuel’s group at EPFL turned to chemistry and developed synthetic strategies that allow site-specific introduction of chemical modifications. This approach elucidates the effect of these modifications without needing to identify the key enzymes responsible, and enables the investigation, for the first time, of the “cross-talk” between different modifications on huntingtin.
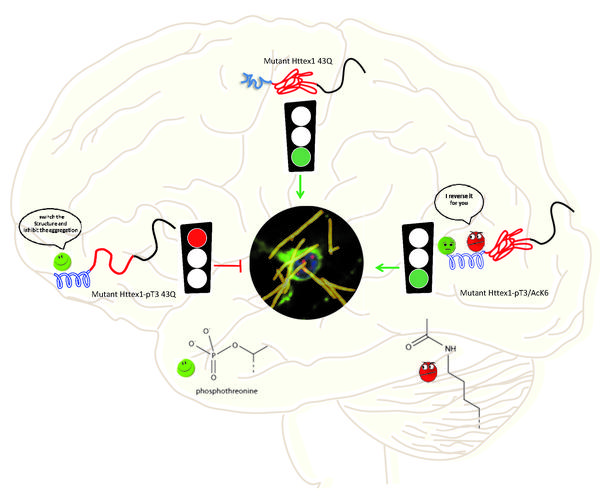
The new strategy combines chemical and bacterial synthesis of proteins to generate a part of mutant huntingtin where many important post-translational modifications take place. This segment is known as “mutant hutingtin exon1” (Httex1) and has been shown to be sufficient for reproducing key features of Huntington’s disease in animal models.
Using the method, the researchers could now generate all the known modified forms of huntingtin in very pure and homogeneous forms. “This allowed us to investigate the effects of post-translational modifications with high precision and under controlled conditions,” says Anass Chiki, the PhD student who led the study.
The researchers explored how these modifications can act as molecular switches that can be exploited to regulate Huntingtin structure, function and toxicity. In this vein, they made three discoveries regarding the relationship between post-translational modifications of Huntingtin and its structure.
First, phosphorylation of threonine on position 3 (T3) stabilizes a helical formation of huntingtin’s first 17 amino acids, and interferes with its ability to aggregate. This could explain why this particular modification is reduced in Huntington’s disease and suggest that modulating the level of this modification could protect against HD disease.
Second, the study found that substituting threonine with another amino acid (glutamate or aspartate) to mimic the charge of the phosphate group, does not fully reproduce the effects of genuine phosphorylation on the structure and aggregation of huntingtin. This is important for biologists who, in the absence of knowledge about the key enzymes that regulate phosphorylation, use this substitution as a go-to solution.
Finally, the scientists were able for the first time to study the impact of acetylation, which takes place on three Lysine amino acids of huntingtin. While acetylation of individual residues did affect huntingtin’s structure or aggregation, acetylation at one lysine (in position 6) did reverse the protective effect of T3 phosphorylation when the two modifications were introduced simultaneously.
“Our work underscores how critical it is to identify the enzymes that regulate huntingtin modifications, both for understanding its biology and unlocking their great potential as viable therapeutic targets for Huntington’s disease,” says Hilal Lashuel. “Being able to generate all disease-modified forms with high purity paves the way for assessing whether these modifications could serve as disease biomarkers for early diagnosis and monitoring disease progression”.
This work was conducted in collaboration with the group of Prof. Giovanni Dietler and Annalisa Pastore and received contributions by the MRC National Institute for Medical Research (London). It was funded primarily by the CHDI foundation and the Swiss National Science Foundation (SNSF).
Reference
Anass Chiki, Sean M. DeGuire, Francesco S. Ruggeri, Domenico Sanfelice, Annalisa Ansaloni, Zhe-Ming Wang, Urszula Cendrowska, Ritwik Burai, Sophie Vieweg, Annalisa Pastore, Giovanni Dietler, Hilal A. Lashuel. Mutant exon1 Huntingtin aggregation is regulated by T3 phosphorylation-induced structural changes and crosstalk between T3 phosphorylaxtion and acetylation at K6.Angewandte Chemie. DOI: 10.1002/anie.201611750