A new approach to fighting chronic myeloid leukemia

A blood smear © 2014 Oliver Hantschel/EPFL
Chronic myeloid leukemia is caused by the hyperactivity of a mutated enzyme. EPFL scientists have discovered an indirect way to regulate this enzyme that could prove more effective than current therapies.
Chronic myeloid leukemia develops when a gene mutates and causes an enzyme to become hyperactive, causing blood-forming stem cells in the bone marrow to grow rapidly into abnormal cells. The enzyme, Abl-kinase, is a member of the “kinase” family of enzymes, which serve as an "on" or "off" switch for many functions in our cells. In chronic myeloid leukemia, the hyperactive Abl-kinase is targeted with drugs that bind to a specific part of the enzyme and block it, aiming to ultimately kill the fast-growing cancer cell. However, treatments are often limited by the fact that the cancer cells can adapt to resist drugs. EPFL scientists have identified an alternative part of Abl-kinase to which drugs can bind and act with a reduced risk of drug resistance. Their work is published in Nature Communications.
Abl-kinase and leukemia
Abl-kinase can turn “on” molecules that are involved in many cell functions including cell growth. In chronic myeloid leukemia, the chromosome that contains the gene for Abl-kinase swaps a section with another chromosome, causing what is known as the “Philadelphia chromosome”. When this mutation takes place in the blood stem cells in the bone marrow, Abl-kinase fuses with another protein, turning into a deregulated, hyperactive enzyme. This causes large numbers of blood-forming stem cells to grow into an abnormal type of white blood cell, which gives rise to chronic myeloid leukemia.
To treat this type of leukemia we use drugs that specifically bind and block a part of Abl-kinase called the “active site”. As the name suggests, this is the part of the enzyme that binds molecules to turn them on. Therefore, blocking the active site with a drug stops the hyperactivity of Abl-kinase caused by the Philadelphia mutation and slow down or even abolishes the production of abnormal cancerous blood cells. The problem is that targeting the active site of Abl-kinase often causes the cancer cells to adapt and develop drug resistance, making them harder to kill.
An indirect path against resistance
A team of researchers led by Oliver Hantschel at EPFL (ISREC) has now discovered a new way to indirectly inhibit the activity of Abl-kinase. The scientists systematically made small, strategic mutations to Abl-kinase that caused its 3D structure to change. Then they tested each mutant version of the enzyme to see if its function would change.
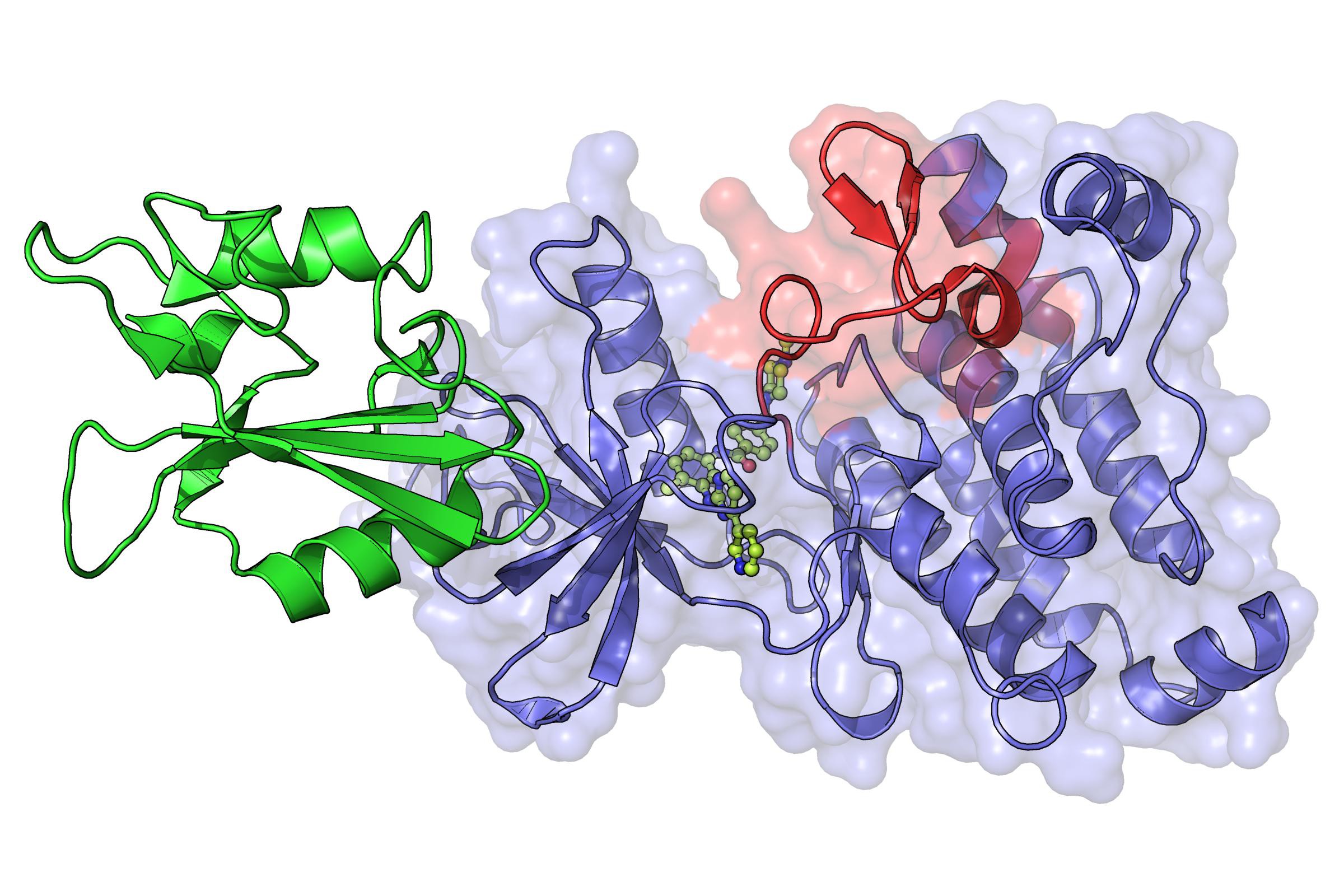
Hantschel’s team built on previous studies showing that Abl-kinase is indirectly controlled by another part of itself called the “SH2 region”, which is located close to the active site. Normally, the SH2 region regulates the active site by opening and closing it. But under the Philadelphia mutation, that regulation is lost. What the scientists discovered was that when the Philadelphia mutation takes effect, the SH2 region actually “clamps” open the active site of Abl-kinase and forces it to go into overdrive.
The discovery provides a first-ever picture of the molecular events surrounding the hyperactivity of Abl-kinase. By blocking the SH2 region, it is possible to modulate the activity of the enzyme, and perhaps stop the growth of leukemic tumors. And since because the SH2 region is common to other kinases, it is likely that effect could extend to other types of cancers as well, particularly those characterized by abnormal kinase activity. Finally, Oliver Hantschel expects that this approach could overcome the problem of tumor drug resistance, as it might offer an alternative way to inhibit the enzyme and mutations of rapidly growing tumor cells may be less likely to occur.
This work was supported by the ISREC Foundation and the Swiss Cancer League.
Reference
Lamontanara AJ, Georgeon S, Tria G, Svergun DI, Hantschel O. The SH2 domain of Abl kinases regulates kinase autophosphorylation by controlling activation loop accessibility.Nat. Comm. DOI: NCOMMS6470
Images to download